

内源性锂或是阿尔茨海默病的关键因素?《Nature》研究揭示新机制与治疗潜力
阿尔茨海默病(AD)的病因研究不仅局限于淀粉样斑块和神经纤维缠结,大脑中微量元素稳态失衡也逐渐成为科学家关注的核心病理特征之一。既往研究多聚焦于金属离子的毒性作用:铜与锌可直接催化Aβ聚集沉淀,并在氧化应激中加速神经元损伤和突触功能障碍;而脑内铁的异常蓄积则通过诱发广泛氧化应激,导致脂质、蛋白质及DNA损伤,成为推动神经元死亡的重要因素。
长期以来,AD的金属组学研究主要集中于金属毒性,研究策略多聚焦于如何螯合或清除有害金属。然而,越来越多证据表明,大脑中的金属稳态是一种精密的动态平衡,金属离子在神经信号传递、能量代谢和抗氧化防御等关键过程中扮演着不可或缺的角色。简单清除金属可能扰乱脑内微环境,导致新的功能紊乱。因此,科学界需要转向一个更具建设性的问题:是否存在某种在AD早期即发生缺失、且对神经系统具有保护作用的必需微量元素?其缺失本身可能正是疾病发生的根源之一。
2025年8月,《Nature》杂志发表了一项突破性研究,将焦点从传统毒性金属转向了一个在精神病学中广泛应用但在AD领域长期被忽视的微量元素——锂。该研究通过单细胞测序、同位素示踪与多组学交叉验证证实,脑内源性锂的轻微减少,足以触发β-淀粉样蛋白聚集、tau蛋白过度磷酸化与突触功能受损的多重病理变化;而外源性补充特定形态的锂,可在小鼠模型中逆转认知损伤,并使突触密度恢复至接近正常水平。这一发现揭示了内源性锂在脑衰老与AD发病中的核心保护作用,不仅增进了对AD病理的理解,更指出了一个潜在的预防与治疗方向。
人类大脑中的锂缺失:从轻度认知障碍到AD
研究者首先使用电感耦合等离子体质谱(ICP-MS)的方法对无认知障碍(NCI)的老年人、轻度认知障碍(MCI,AD的前驱期)或AD患者的大脑和血液进行了系统的金属组学分析。在检测的27种金属中,唯有锂(Li)在MCI和AD患者的大脑前额叶皮层中均发生显著降低;并且这一发现在两个独立人群中得到重复(图1a-e)。这种下降并非源于全身性锂水平变化,而是大脑局部锂稳态的特异性破坏。
更关键的是,研究发现,AD病理的核心特征——β淀粉样蛋白(Aβ)斑块,竟是一个锂陷阱。通过激光剥蚀-电感耦合等离子体质谱技术,研究人员量化了Aβ斑块中的Li与额叶皮层的无斑块区域的比较,结果显示锂在Aβ斑块中高度富集。这意味着,随着病程进展,越来越多的锂被锁在斑块中,导致其在功能性脑区(非斑块区域)的生物利用度下降,从而可能加剧神经毒性(图1f-g)。
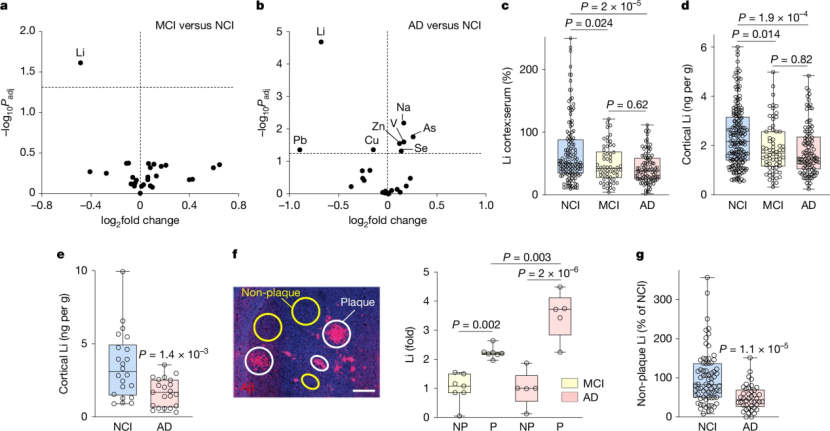
图1. MCI和AD患者中,脑中内源性锂稳态受到干扰
锂缺失在小鼠模型中的因果作用
为证实锂缺失的因果作用,研究团队构建了锂缺乏饮食的小鼠模型。结果显示,通过给予小鼠锂去除的饮食将大脑皮层锂水平降低约50%后,无论是AD模型小鼠3xTG或J20小鼠模型,还是正常衰老的野生型小鼠,都出现了全方位的病理加速:Aβ沉积显著增加;磷酸化Tau蛋白累积加剧,形成类似神经原纤维缠结的结构;小胶质细胞被异常激活,呈现促炎状态以及突触、轴突和髓鞘大量丢失(图2a-f)。而在水迷宫和新物体识别等学习认知能力的测试中,这些小鼠的学习与记忆能力也出现了严重受损,甚至在正常衰老的野生型小鼠中,单纯缺锂就足以导致记忆功能下降;而它们的运动能力、视觉感知等其他功能并未受损(图2g-o)。这表明,内源性锂对维持正常的认知功能至关重要。
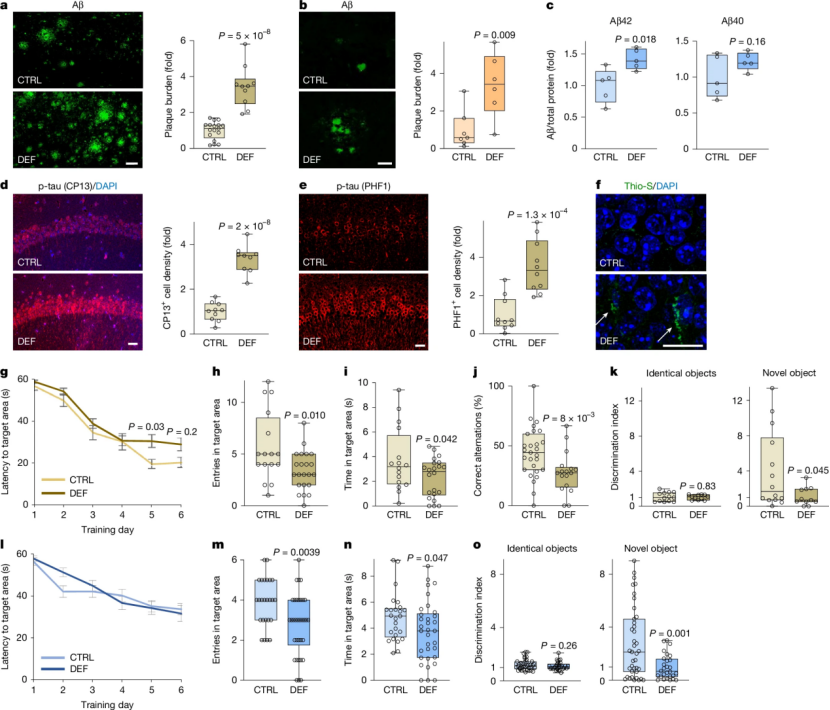
图2. 锂缺失加速AD病理和认知能力下降
锂缺失对全细胞转录组的影响
研究人员进一步通过单细胞测序的手段对锂缺乏3xTg小鼠的海马进行“细胞级普查”,研究发现兴奋性神经元、少突胶质细胞和小胶质等7大类型细胞的转录组发生了显著的变化:兴奋性神经元中与突触信号传导和结构相关的基因(如Homer1, Grik1等)普遍下调,而氧化应激等与神经退行性病变相关的通路被激活;少突胶质细胞中负责轴突包裹和髓鞘形成的基因(如Mbp, Mag等)则显著下调(图3a-d);锂缺失与活体人脑早期Aβ或Aβ/tau活检样本的DEG高度重叠(图3e)。同时,这些转录组的变化也在功能上得到了验证,锂缺乏导致了树突棘的丢失、突触蛋白(如PSD-95和突触素)的减少、髓鞘的破坏以及少突胶质细胞和轴突数量的下降(图3f),提示内源性锂的缺失会引发多种脑细胞(特别是神经元和少突胶质细胞)的基因表达谱发生病理性改变,这些改变直接导致了突触功能和髓鞘结构的损害,从分子和细胞层面解释了锂缺乏为何会加速认知衰退,为“锂是生理守门员”提供了基因表达层面的直接证据。
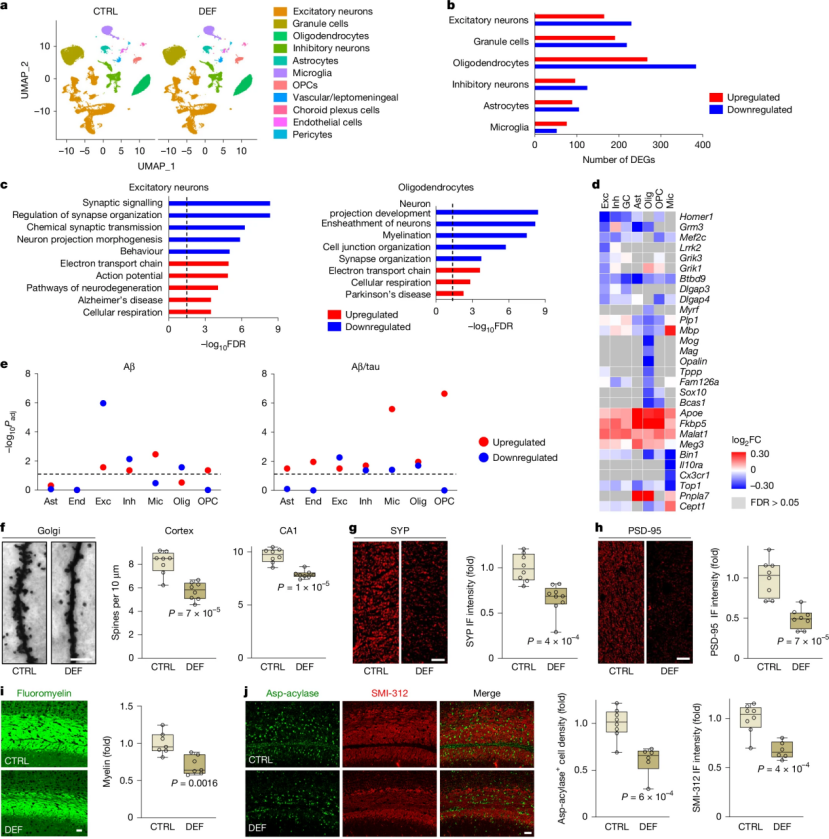
图3. 内源性锂缺失诱发多种细胞类型转录组失调
分子机制探索——GSK3β是关键中介
锂缺失究竟如何引发如此广泛的破坏?研究人员通过使用差异化表达基因(DEG)的 Ingenuity/IPA 网络分析确定了因Li缺乏而改变的信号通路,结果显示Wnt-β-连环蛋白信号传导受到显著影响,预计在小胶质细胞、兴奋性神经元和少突胶质细胞中受到抑制。Wnt-β-连环蛋白信号传导的中心调节因子是糖原合成酶激酶3β(GSK3β),GSK3β是AD中的一个关键致病激酶,它能促进Tau蛋白磷酸化和Aβ的产生,而锂是GSK3β的天然抑制剂。研究发现在锂缺乏的大脑中,海马CA1神经元,少突胶质细胞和小胶质细胞中的GSK3β的表达和活性均显著升高。而使用GSK3β抑制剂,CHIR99021和PF-04802367,均可以逆转由锂缺失引起的大部分病理变化,包括Aβ沉积、Tau磷酸化、髓鞘损伤和神经炎症(图4)。这表明,GSK3β的激活是锂缺失下游的核心事件。
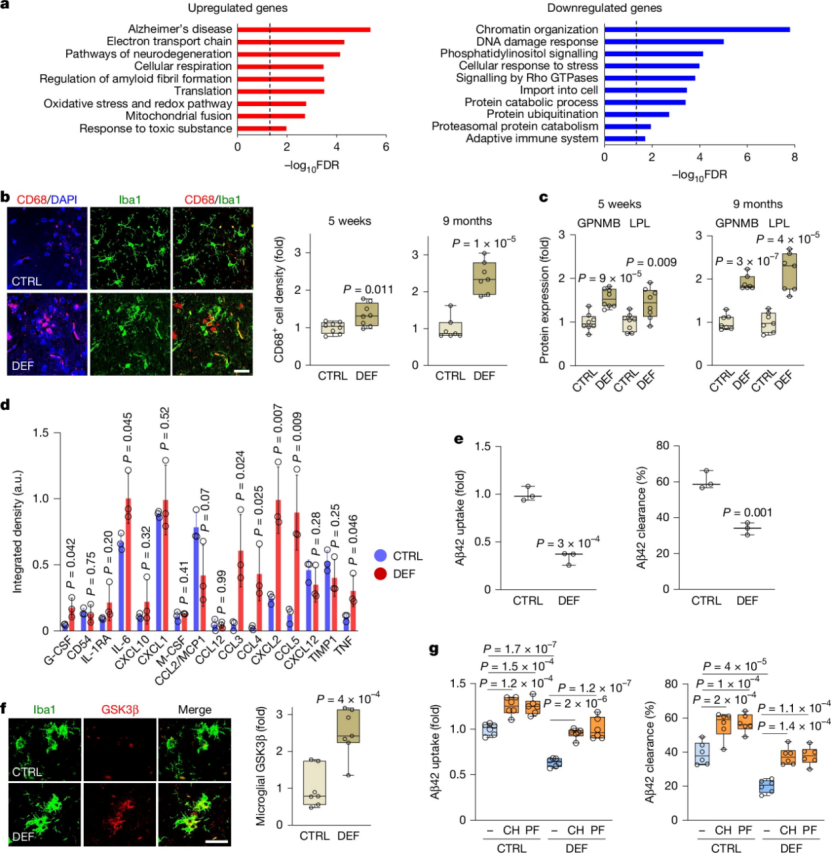
图4. GSK3β激活导致与锂缺失相关的病理变化
创新治疗策略:避免斑块结合的新型锂盐
鉴于锂容易被Aβ斑块结合,常规补锂策略可能效果有限。研究团队创新性地筛选了多种锂盐,发现临床常用的碳酸锂与Aβ结合力强,补充后仍会大量被困在斑块中。而一种名为乳清酸锂的有机锂盐,其电导率较低,与Aβ的结合亲和力显著弱于碳酸锂。
在AD小鼠模型中,低剂量乳清酸锂能够有效提升非斑块区域的锂水平,显著减少Aβ斑块和磷酸化Tau的累积,抑制神经炎症,保护突触和髓鞘结构,并几乎完全逆转记忆丧失(图5)。相比之下,同等剂量的碳酸锂效果有限。长期低剂量乳清酸锂治疗在衰老小鼠中未观察到肾脏或甲状腺毒性,展现了良好的安全性。
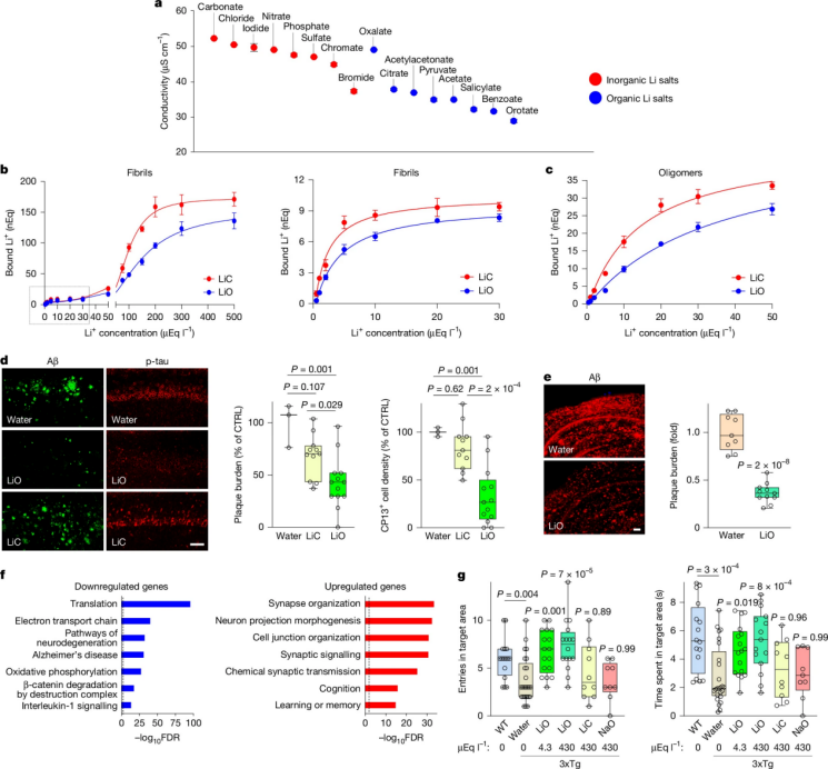
图5. “绕开”斑块的锂盐治疗可有效改善AD相关表型
前景与展望
这项研究首次系统揭示了内源性锂作为一种重要的生理性神经保护因子,在维持大脑健康、抵御衰老和AD相关病理中的核心作用。基于这一发现,研究提出了一个AD发病的循环模型:Aβ沉积导致局部锂缺失,锂缺失进而激活GSK3β激酶、损害小胶质细胞的清除功能,从而加剧Aβ与Tau病理积累,进一步破坏锂稳态,形成病理级联反应。
该模型表明,大脑锂稳态的扰动发生在MCI甚至更早阶段。这提出了一个可能性:能否通过检测体液中的锂水平或利用新型影像学技术,实现对锂失衡的早期识别?如果可行,锂缺失有望成为一个全新的生物标志物,用于早期筛查和风险评估,从而实现超早期诊断。
除了诊断价值,这项研究也为AD防治带来了新视角。以乳清酸锂为代表的避免斑块结合的锂盐,展现出在AD预防与早期干预中的重要潜力。未来药物开发可能转向生理性补充与稳态重建的新范式,开发生物利用度更高、与Aβ结合更弱、安全性更优的新型锂制剂。结合个体锂水平、Aβ负荷及遗传背景的精准补锂方案,可能成为神经退行性疾病个性化医疗的新方向。
此外,这一发现还为群体预防开辟了可能路径。如同食盐加碘预防甲状腺疾病,未来或可探索通过饮用水或膳食补充特定形态的锂,实现对广泛人群认知健康的维护。这种基于营养干预的一级预防策略,特别适用于AD高危人群,为在公共卫生层面延缓或阻止疾病发生提供了新的可能性。
参考文献
Aron L, Ngian ZK, Qiu C, et al. Lithium deficiency and the onset of Alzheimer's disease. Nature. 2025.